
Research Article | DOI: https://doi.org/BRCA-RA-24-006
‘’Targeting Cellular Hypoxia in βcells and Autophagy in Prevention and Propagation of Type 2 Diabetes-A narrative review’’
Abstract
portrays a chronic disease possessing considerable hyperglycemia; dysfunctional insulin liberation by pancreatic βcells is an emblem of such disease. Recent studies have illustrated that hypoxia takes place in the pancreatic βcells of patients having T2D with hypoxia as a result leading to abnormalities of insulin liberation in addition to elimination of βcell mass via mechanistic modes inclusive of activation of hypoxia inducible factor alpha(HIF 1-α), induction of transcriptional suppressors along with activation of 5’ AMP-activated protein kinase(AMPK) . Earlier we had reviewed on the aetiopathogenesis of Type 2 diabetes mellitus(T2D) along with role of gutmicrobiota(GM),diabesity,oral health AGEs stimulated & ERand Inflammatory Stress- modulated control of the GLUT4 expression (SLC2A4) promoted genes,;details of epigenetics, mitochondrial melatonergic pathwaysand different methods of use of various plant products ,role of extracellular vesicles ,iron&mineral metabolism and umpteen other articles Here our concentration is on insight into βcell hypoxia that might result in dysfunctional insulin liberation in T2DM .An understanding of βcell hypoxia might aid in generation of innovative strategies for the treatment of T2DM . Further with emerging evidence of how autophagy might be implicated in propagation of Type 2 Diabetes,thereby targeting both hypoxia and autophagy might bethe mechanistic modes of how separate plant products are contributing in T2D avoidance as well as propagation.Here we have attempted to give insight regarding how βcells hypoxia aids in generation of βcells impairment in T2D. Achieving greater insight of βcell hypoxia might aid in generating innovative approaches for T2D treatment.
Introduction:
Diabetes mellitus( DM)¬Ý represents a chronic¬Ý disorder associated with¬Ý considerable hyperglycemia and portrays one of the commonest etiological factor of mortality along with morbidity globally¬Ý .It has been determined that 529 million¬Ý people have been¬Ý living with DM worldover of which Type2 Diabetes mellitus( T2DM)¬Ý was implicated in 96% of full patients, in addition to proportion of the patients with DM have been estimated to escalate greater than double of 1.3 billion¬Ý individuals globally by year 2050[1]. Etiological factors responsible for T2DM are¬Ýcomplicated ¬Ýcrosstalking amongst numerous genetic as well as¬Ý environmental factors. The genetic makeup results in insulin resistance¬Ý (IR) along with pancreatic Œ≤cells, whereas escalated weight along with sedentary life aggravates such metabolic abnormalities[2]. Dysfunctional insulin liberation in addition to IR specialized properties of T2DM[2]. In case of IR pancreatic Œ≤cells escalate insulin liberation regarding sustenance of normal glucose tolerance; nevertheless, once Œ≤cells lose their capability of¬Ý escalating¬Ýinsulin liberation the plasma quantities of glucose get escalated. Continued¬Ý exposure to hyperglycemia possess inimical sequelae over Œ≤cell numbers along with working;a postulate¬Ý ¬Ýreferred to as¬Ý gluotoxicity which results in the generation as well as propagation of T2DM[3,4]. Hyperglycemia has a¬Ý negative impacts via plethora of mechanistic¬Ý modes¬Ý toxic¬Ýactions¬Ý inclusive of Oxidative¬Ý ¬Ýstress( OS) along with endoplasmic reticulum (ER) stress in addition to inflammation[5]. Nevertheless, recent studies have¬Ý ¬Ýpointed¬Ý that hyperglycemia further stimulates hypoxia in Œ≤cells[6,7]. Hypoxia in turn¬Ý aids in Œ≤cell impairment through various mechanistic¬Ýmodes¬Ý ¬Ýinclusive of hypoxia inducible factor¬Ý ¬Ýalpha(HIF 1-Œ±) [8]. Earlier we¬Ý ¬Ýhad reviewed on the aetiopathogenesis of¬Ý ¬ÝType 2 diabetes mellitus(T2D) along with role of gutmicrobiota(GM),diabesity,oral health AGEs Stimulated & ERand Inflammatory Stress- Modulated Control of the GLUT4 [removed]SLC2A4 promotedgenes,;details of epigenetics, mitochondrial melatonergic pathwaysand different methods of use of various plant products ,role of ecv,iron&mineral metabolismand umpteen other articles [9-25].Here our¬Ý concentration is¬Ý ¬Ýon insight into Œ≤cell hypoxia that might result¬Ý in dysfunctional insulin liberation in T2DM .Aninsight of Œ≤cell hypoxia might¬Ý aid in generation of innovative strategies for the treatment¬Ý of T2DM .¬Ý
Methods
Thus a narrative¬Ýreview was carried out using the pubmed, Web of Science , Medline, Embase, Cochrane reviews,¬Ý and Google Scholar, Search¬Ý engine with the MeSH Terms; Type 2 diabetes mellitus(T2D); Hypoxia; the mitochondrial melatonergic pathways; oxidative¬Ý ¬Ýphosphorylation (OXPHOS); adenosine triphosphate(ATP); insulin exocytosis; hyperglycemia; Pancreatic Œ≤cells; prolyl hydroxylase domain(PHD) proteins ; HIFs from 1995 till date in 2024.
Results
We found a total of 250 articles ,out of which we selected 68 articles for this review.No meta-analysis was done.¬Ý
2. Stimulation of Hypoxia in pancreatic Œ≤cells by hyperglycemia¬Ý
In case of¬Ýnormoxic IR pancreatic Œ≤cells, glucose gets metabolized into pyruvate¬Ý ¬Ýthrough¬Ý ¬Ý glycolysis in addition to its further oxidation takes place for¬Ý generation of¬Ý adenosine triphosphate(ATP) through oxidative¬Ý ¬Ýphosphorylation (OXPHOS).An¬Ý escalation of ATP results in the¬Ý closing of¬ÝATP sensitive potassium¬Ý (KATP ) channels In pancreatic Œ≤cells resulting in membrane¬Ý depolarization, Calcium(Ca2+ ) influx as well as insulin¬Ý vesicle exocytosis[26]. Cellular¬Ý ¬Ýoxygen¬Ý quantities are¬Ý controlled ¬Ýby the harmony amongst supply along with requirement of oxygen in addition to once hypoxia takes place oxygen utilization is greater than its supply . Acknowledged the considerable requirement of mitochondrial OXPHOS at the time of insulin liberation, Œ≤cells¬Ý utilize considerable oxygen¬Ý quantities . Actually¬Ý it has been revealed by¬Ý the group of¬ÝYamagata¬Ý et al.[¬Ý [6,7,27], along with others that pancreatic islets of Langerhans¬Ý ¬Ý as well as¬Ý Œ≤cells lines become¬Ý hypoxic [6,7],¬Ý with ease under escalated¬Ý glucose¬Ýsituation [6,7,27]. Such¬Ý studies have further illustrated that islets in animal models¬Ý of T2DM are hypoxic[28]. Thereby inadequate oxygen supply might further be implicated in Œ≤cells hypoxia in vivo.
The oxygen tension in¬Ý ¬Ýmaximum mammalian cells is¬Ý ¬Ývarying amongst the¬Ý values¬Ýof 20-65 mmHg(parallel to 3-9%O2) [29],¬Ýas well as the¬Ý average tissue oxygen tension at the surface of the normal mouse islets varying amongst the¬Ý values¬Ý of44.7-45.7mmHg (parallel to 6.3-6.4%O2) [30]. Hypoxic reactions have been illustrated to take place in culture situations in vitro [31]. Continuous exposure of MIN6 Œ≤cells to 5%O2 tension result in cellular hypoxia with dysfunctional insulin liberation along with hampers Œ≤cells growth ; 3%O2 tension resulted¬Ýin apoptosis with ease in addition to¬Ýdiminished¬Ý Œ≤cells numbers as well as working [32,33]. Thereby hypoxic stress represents the mechanistic¬Ý ¬Ýmode behind Œ≤cells failure in case of T2DM[32,34]., [32reviewed in ref 35].(Figure1)
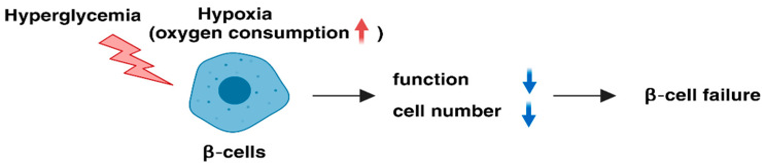
3. Part of HIFs in Pancreatic Œ≤cells¬Ý
The sustenance of oxygen homeostasis¬Ý is significant¬Ý in reference to ATP generation in addition to energy¬Ý accessiblity in cells .Thereby all mammals possess the¬Ý capacity of sensing, reacting to as well as rectifyingy hypoxia .HIFs portray crucial members of the¬Ý basic-loop-helix PerArntSim¬Ý transcription factor family along with are comprised of oxygen sensitive HIF 1-Œ± subunit in addition to¬Ý a HIF 1Œ≤/ aryl hydrocarbon receptor nuclear translocator (ARNT) subunit which gets¬Ýconstitutively¬Ý expressed¬Ý [31,36,].Three¬Ý kinds of HIFs are existent (HIF 1-Œ±, HIF 2-Œ±, in addition to HIF 3-Œ±); nevertheless, maximum of the transcriptional reactions are apparently secondary to HIF 1-Œ±, along with HIF 2-Œ±[37].At the time of normoxic situations, HIF 1-Œ± undergoes hydroxylation at the 2proline residues amongst the oxygen based breakdown¬Ý domain by the prolyl hydroxylase domain(PHD) proteins in the¬Ýexistence of oxygen,2 oxoglutarate as well as iron . Hydroxylated, HIF 1-Œ± subunits get poly ubiquitinated by the vonHippel-Lindau protein in addition to are targeted for¬Ý proteasomal breakdown. Hydroxylation by the PHD proteins gets avoided along with¬Ý following breakdown in case of hypoxic situations . In view of¬Ý this¬Ý stabilized HIF 1-Œ± undergoes dimerization with HIF 1Œ≤ along with activation of a substantially greater quantities of target genes inclusive of those implicated in glycolysis ,erythropoiesis , in addition to angiogenesis by binding to the hypoxia response element in¬Ý their promoter areas.
Three¬Ýkinds of PHD proteins(PHD1, PHD2 in addition to PHD 3)get¬Ý expressed in Œ≤cells[38], as well as HIF 1-Œ± gets brokendown pacily in case of normal oxygen situations .Nevertheless, HIF 1-Œ± is existent in normoxic¬Ý Œ≤cells[39]. Glucose¬Ý transporter 2¬Ý (GLUT2) portrays a lesser affinity glucose¬Ý transporter whose requirement is for sustenance of normal glucose stimulated insulin liberation in Œ≤cells[40]. Glucokinase,that is a rate¬Ý restricting glycolytic enzyme,¬Ý works in the form of a sensor for the physiological insulin liberation in Œ≤cells[41]. Intriguingly elimination of¬Ý Hif-Œ± gene in Œ≤cells results in dysfunctional insulin liberation in addition to glucose intolerance in¬Ý ¬Ýmice¬Ýhaving a diminished expression of soluble¬Ý carrier family¬Ý 2¬Ýmember 2 (Slc2a2)gene that encodes GLUT2 along with Gck¬Ý gene( that encoded glucokinase) [39]. Frequently HIF 1-Œ± knockout (KO) diminished¬Ýexpression quantities of Slc2a2 in addition to¬Ý Gck¬Ýare considerably¬Ý repressedin case of¬Ý insulin liberation in¬Ý ¬ÝMIN6 Œ≤cells at the time of¬Ý normoxic situations [39].¬Ý Thereby HIF 1¬Ýexpression¬Ý at basal¬Ý ¬Ýquantities for¬Ý insulin liberation is necessary, despite mechanistic¬Ý modes behind this diminished¬Ý expression quantities of Slc2a2 in addition to¬ÝGckbyHIF1-Œ±insufficiencyareuncharted(Figure2A,B).
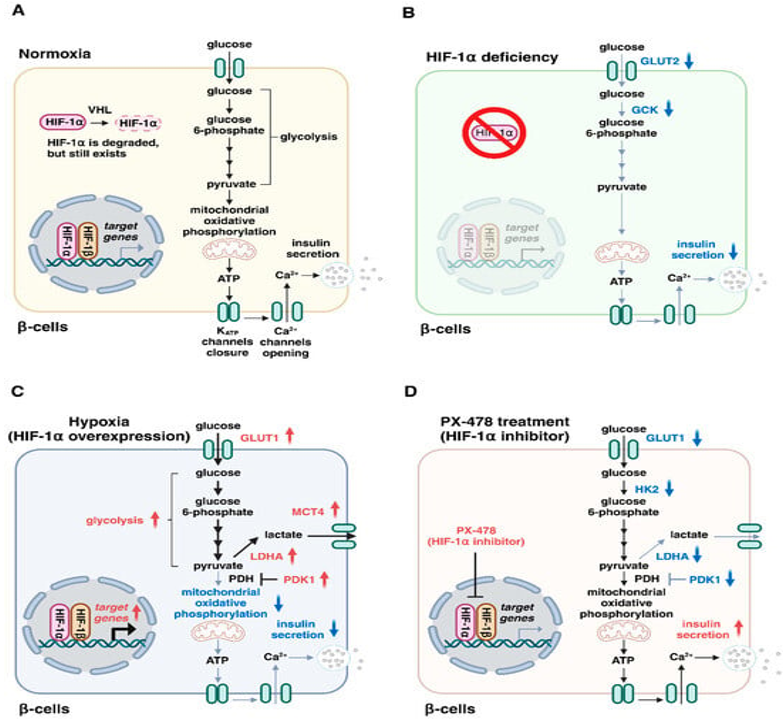
Legend for Figure 2.¬Ý
Courtesy ref no-35-Roles of hypoxia-inducible factor (HIF)-1 in insulin secretion by β-cells. (A) Glucose is metabolized via the glycolytic pathway and mitochondrial oxidative phosphorylation, resulting in the generation of adenosine triphosphate (ATP), KATP channel closure, Ca2+ entry, and insulin exocytosis. Under normoxic conditions, HIF-1α is degraded by von Hippel–Lindau (VHL) proteins. (B) HIF-1α is degraded under normal oxygen conditions, but remains present in normoxic β-cells. HIF-1α deficiency causes impaired insulin secretion with a decreased expression of glucose transporter type 2 (GLUT2) and glucokinase (GCK). (C) HIF-1α overexpression switches glucose metabolism from mitochondrial oxidation to glycolysis, thereby leading to the attenuation of mitochondrial activity and impaired insulin secretion. (D) Treatment with the HIF-1α inhibitor PX-478 prevents the upregulation of HIF-1α targets (GLUT1, HK2, LDHA, and PDK1) and restores insulin secretion in metabolic workload. HK2, hexokinase 2; LDHA, lactate dehydrogenase A; MCT4, monocarboxylate transporter 4; PDH, pyruvate dehydrogenase; PDK1, pyruvate dehydrogenase kinase 1.
Additionally,¬Ý ¬Ý HIF 1-Œ± confers protection against Œ≤cells damage¬Ý in type 1 diabetes mellitus(the autoimmune kinds of diabetes) [42]. Furthermore HIF 1-Œ±/ ARNT insufficiency further repressed insulin liberation in Œ≤cells[43].¬ÝInterestingly, declined HIF 1-Œ± in addition to ARNT /HIF1Œ≤ have been found in T2DM patients [39,43]. Moreover HIF 1-Œ± signaling gets repressed in a complicated¬Ý manner by hyperglycemia via PHD) proteins based mechanistic¬Ýmodes[8,44].¬Ý Such findings robustly portray that HIF 1-Œ± proteins possess¬Ýa significant¬Ý ¬Ýpart in sustenance of Œ≤cells working as well as the¬Ýmanner¬Ý dysfunctional HIF 1 signaling is¬Ý responsible for Œ≤cells impairment in type2 diabetics .¬Ý
Compared to that it have further been illustrated that HIF 1-Œ± expression is escalated in the Œ≤cells¬Ý ¬Ýof various diabetic animals, inclusive of ob/ob¬Ý ¬Ýmice, mice which received high¬Ý fat diet(HFD), in addition to db/db mice [7,45]. Maintainance of HIF 1-Œ± overexpression by the elimination of¬Ý Vhl gene(implicated in encoding¬Ý ¬ÝvonHippel-Lindau protein ) results¬Ý in dysfunctional insulin¬Ý ¬Ýliberation along with glucose intolerance in¬Ý ¬Ýmice[46], pointing that¬Ý the upregulation¬Ý ¬Ýof¬ÝHIF 1-Œ± is inimical for the working of Œ≤cells as well as aids in T2DM generation. HIF 1-Œ± results¬Ý in the activation¬Ý ¬Ýof the transcription of the genes¬Ý encoding¬Ý ¬ÝGLUT 1, glycolytic enzymes(glucose-6- phosphatase¬Ý isomerase, and phosphoglycerate mutase)ii) pyruvate dehydrogenase kinase (PDK) iii) lactate dehydrogenase A(LDHA) in addition to iv) monocarboxylase transporter 1(MCT4) [47], PDK 1 is involved in the inactivation of the¬Ý enzyme pyruvate dehydrogenase which¬Ý is responsible for the transformation¬Ý of¬Ý ¬Ýpyruvate to acetylCoA for the mitochondrial tricarboxylic acid(TCA)/Krebs Cycle. LDHA prevents pyruvate from gaining entry in to TCA by¬Ý ¬Ýtransforming pyruvate to lactate¬Ý in addition to MCT4 facilitates extrusion¬Ý ¬Ýof¬Ý lactate from cells. Sequentially, the major influence of HIF 1-Œ± on glucose metabolism is¬Ýswitching energy metabolism from mitochondrial respiration to glycolysis. Nevertheless, mitochondrial oxidative metabolism possesses key¬Ý part in the regulation of insulin¬Ý ¬Ýliberation[48]. the main exposition for the inimical actions in reference to HIF 1-Œ± on insulin¬Ý ¬Ýliberation¬Ýis amelioration of mitochondrial actions(Figure2C). Interestingly,¬Ý therapy¬Ýof¬Ý diabetic mice by utility of HIF 1 hampering agent PX-478¬Ýresults¬Ý in improvement¬Ý of insulin¬Ý liberation along with glucose intolerance[45], indicating that hampering HIF 1-Œ± might be¬Ý a plausible treatment for type2 diabetics(Figure2D). Overall such outcomes point that a harmonious in addition to sufficient quantities of HIF 1-Œ± actions¬Ý is imperative for the normal insulin¬Ý ¬Ýliberation by pancreatic Œ≤cells.¬Ý
Compared to that¬Ý HIF 2-Œ± a paralog(created by gene duplication) of HIF 1-Œ± further undergoes dimerization with HIF 1Œ≤ for the activation of target genes¬Ý in reaction¬Ý to hypoxia. Nevertheless, HIF 1-Œ± as well as HIF 2-Œ± possess unique Part in Œ≤cells .The manner described earlier, Œ≤cells particular Hif 1-Œ± KO¬Ý mice¬Ý illustrate dysfunctional¬Ý insulin¬Ý liberation along with glucose intolerance[39] Compared to that HIF 2-Œ± insufficiency in Œ≤cells does not lead to dysfunctional¬Ý insulin¬Ý liberation along with glucose intolerance in mice¬Ý getting normal chow diet[48]. A chronic escalation in mitochondrial metabolism¬Ý ¬Ýescalates electron flux in the¬Ýelectron transport chain(ETC) leading¬Ýto escalated generation of reactive oxygen species(ROS) [49]. HIF 2-Œ± possesses significant part in the controlling of the cellular redox status¬Ý ¬Ýby activation of the antioxidant gene expression Sod2 (encoding superoxide dismutase), as well as Cat(encoding catalase) in addition to confers protection against mitochondrial injury by ROS[50]. Frequently there is¬Ý reduced expression of the antioxidant genes¬Ý in the islets of Œ≤cells particular Hif 1-Œ± KO mice along with such mice form dysfunctional¬Ý insulin liberation along with glucose intolerance on getting¬ÝHFD[49]. Such outcomes point that HIF 2-Œ±¬Ý is involved in preservation of Œ≤cells working¬Ý in situations of metabolic excess by stimulating¬Ý generation of antioxidant genes expression.¬Ý
4. Part of Transcriptional Suppressors in Hypoxic Œ≤cells¬Ý
Working of the HIFs¬Ý ¬Ýis¬Ýbasically in the form of activators of transcription ; nevertheless, suppression of transcription further takes place for hampering events which possess considerable energy requirement in case of hypoxic situations[51]. Actually, 5% of genes inclusive of certain genes implicated in insulin liberation¬Ý caused downregulation in hypoxic islets along with MIN6 Œ≤cells [32,33,52], pointing that genes suppression portrays one more adaptive reaction to hypoxia in Œ≤cells. Global¬Ý gene expression¬Ý evaluation displayed that basic- helix -loop-helix family¬Ý member E40 (BHLHE40) in addition to activating transcription factor 3(ATF3) represent hypoxia stimulated transcriptional suppressors in Hypoxic Œ≤cells (Figure3) [33].
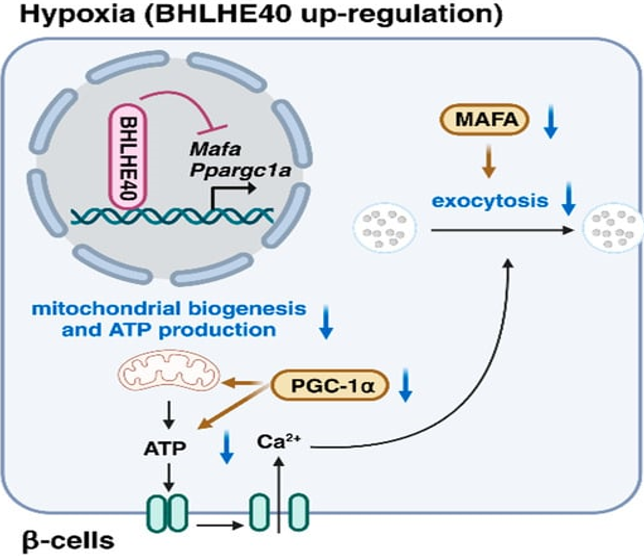
Legend for Figure 3.¬Ý
Courtesy ref no-35-The transcriptional repressor basic helix-loop-helix family member E40 (BHLHE40) is highly induced in hypoxic β-cells. BHLHE40 inhibits insulin secretion by suppressing the expression of musculoaponeurotic fibrosarcoma oncogene family A (MAFA), a transcription factor that regulates insulin exocytosis, and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α), which plays important roles in mitochondrial biogenesis and adenosine triphosphate (ATP) production.
BHLHE40(alias DEC1/SHARP2/STRA13) portrays¬Ý a member of basic- helix -loop-helix family¬Ý in addition to works by binding to DNA at the¬Ý ¬ÝClass EB motifs[53]. The¬Ý transcription factor musculoaponeurotic fibrosarcoma oncogene(MAFA) which possesses a key part¬Ý in glucose stimulated insulin liberation,by¬Ý controlling gene implicated in insulin exocytosis inclusive of Stxbp1( encoding MUNC 18-1) as well as¬Ý Stx1a ( encoding syntaxin A) [55]. Peroxisome Proliferator Activated Receptor Œ≥¬ÝCoactivator -1Œ±(PGC-1Œ±) whose¬Ýencoding gets done by¬Ý Ppargc1Œ± controls mitochondrial biogeneration as well as ATP generation[55]. Remarkable induction of the¬Ý transcriptional suppressor BHLHE40 expression in Œ≤cells by hypoxia along with¬Ý ¬Ýsuppresses insulin liberation by suppressing¬Ý expression of Mafa in addition to Ppargc1Œ±.. Persistently¬ÝŒ≤cells particular Bhlhe40 insufficiency results¬Ý in improvement of insulin¬Ý ¬Ýliberation along with glucose intolerance in ob/ob mice.¬Ý
ATF3 further represses the expression of genes implicated in glucose metabolism inclusive of Ins1( encoding insulin1) Ins2( encoding insulin2) in addition to Irs2( encoding insulin Receptor¬Ý substrate-1(IRS-2)] [33,56]. Additionally, the hypoxia stimulated upregulation of the proinflammatory¬Ý II1b¬Ýas well as proapoptotic¬Ý Noxa genes along with activation of caspase-3¬Ýget suppressed¬Ý by Atf3 insufficiency in MIN6 Œ≤cells [33,56,57].Such observations further point that the transcriptional suppressor ATF3 is implicated in hypoxia stimulated Œ≤cells impairment in addition to elimination.
5. Controlling of Various Stress Pathways in Œ≤cells by Hypoxia¬Ý
5‚Äô adenosine mono phosphate (AMP)-activated protein kinase(AMPK) portrays an evolutionary preserved¬Ý serine /threonine kinase. Activation of AMPK takes place in reaction to energy stresses for instance hypoxia¬Ý by¬Ý sensing escalated quantities of AMP as well as/or adenosine¬Ý di phosphate: ATP ratio by hampering anabolic events which generate ATP[58]. Hepatocyte¬Ýnuclear¬Ý factor 4 alpha (HNF-4Œ±) represents a¬Ý transcription factor from the nuclear receptor super family¬Ý ¬Ýwhich possesses¬Ý significant key part in insulin liberation[59]. It was observed¬Ýby¬Ý the group of¬Ý Yamagataet al.[60], that hypoxia stimulated AMPK activation diminished insulin liberation by diminishing the¬Ý stability of¬ÝHNF-4Œ± [60]. Thereby downregulation of HNF-4Œ± by activation of AMPK might be responsible for the dysfunctional insulin liberation in¬Ý case of¬Ýhypoxic¬Ý situations.
Dysfunctional protein homeostasis(alias¬Ý proteostasis) in the ER¬Ý results¬Ý in accrual of unfolded along with aberrantly¬Ý folded¬Ý protein alias ER stress, resulting¬Ýin¬Ý ¬Ýactivation of the ER unfolded¬Ý ¬Ýprotein responses(UPRER) for the the¬Ý ¬Ýamelioration of proteostasic stress[61]. Hypoxia escalates Œ≤cells demise by hampering the expression of¬Ý adaptive UPRER genes inclusive of Hspa5( encoding heat shock protein family A member 5) Hsp90b1 (encoding heat shock protein90 beta¬Ý family member1)Fkbp11 ( encoding FKBP prolyl isomerase 11) in addition to spliced Xbp1(encoding X-box binding protein 1). Such hampering actions¬Ýof¬Ý hypoxia¬Ý modulated by the activation of c-Jun-N-terminal kinase¬Ý (JNK) as well as DNA damage¬Ý inducible transcripts3 however are autonomous of HIF 1-Œ±[62]. UPRER¬Ýgetting inactivated¬Ý might be the¬Ý ¬Ýcellular mechanistic¬Ý mode behind escalated¬Ý ¬Ýcell demise by hypoxic stress .
OS gets stimulated in tissue in case of escalated glucose situations. Noticeably, Œ≤cells possess considerable susceptibility¬Ý specifically¬Ý to ROS in view of their expression¬Ý of minimal¬Ýquantities¬Ý ¬Ýof antioxidant¬Ý ¬Ýenzymes¬Ý inclusive of glutathione¬Ýperoxidase(GPx), catalase(CAT) , as well as mitochondrial¬Ý manganese SOD¬Ýalong with ROS formed¬Ý in Œ≤cells decline insulin¬Ý genes expression reducing the expression in addition to /or DNA binding actions of pancreatic as well as¬Ý duodenal homeobox 1 (PDX1) transcription facto[63]. Interestingly, hypoxia further¬Ýescalates ROS¬Ý generation at the mitochondrial ETC[64]. Such outcomes robustly¬Ýpoint that hypoxia¬Ý stimulated ROS generation further¬Ý is implicated in Œ≤cell impairment .
From these findings documented above it is clear that hypoxia impacts a¬Ýplethora of¬Ý events cascade of¬Ý at the time of glucose stimulated insulin liberation . Particularly hypoxia ameliorated insulin liberation by switching glucose metabolism from mitochondrial respiration to glycolysis via the activation of HIF 1. Hypoxia further hampered¬Ýinsulin liberation by repressing the expression of MAFA(exocytosis) as well as Peroxisome Proliferator Activated Receptor Œ≥Coactivator -1Œ±(PGC-1Œ±)via the activation of transcriptional suppressor¬ÝBHLHE40 . Additionally, the hypoxia stimulated activation of AMPK resulted¬Ý ¬Ýin downregulation of the¬Ý expression of HNF-4Œ±¬Ý ¬Ýresulting in aberrant insulin liberation. Moreover, hypoxia¬Ý stimulated ROS generation¬Ý hamper insulin gene expression¬Ý via the decontrolling of PDX1(seeFigure4) .
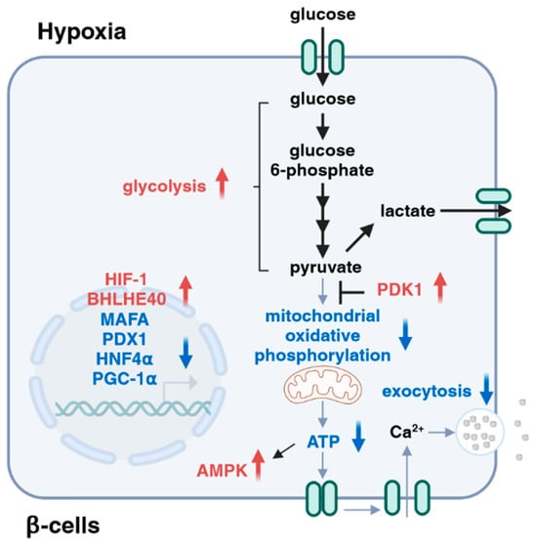
Legend for Figure 4
Courtesy ref no-35-Roles of hypoxia in insulin secretion. Hypoxia affects multiple steps during the processes of glucose-stimulated insulin secretion, including dysregulation of transcription factors (e.g., MAFA, PDX1, and HNF4Œ±), attenuation of mitochondrial activities, activation of AMPK, and inhibition of exocytosis¬Ý
6.Conclusions
Diabetes implicates a clinical scenario where pancreatic Œ≤cells are engulfed in a vicious cycle which leads to a dysfunctional insulin reaction to glucose generated hyperglycemia that¬Ý sees to it that Œ≤cells lose their efficacy in reference to insulin liberation that leads to¬Ýa improvement¬Ý ¬Ýof hyperglycemia that¬Ý causes a minimum of¬Ý part restoration of Œ≤cell working [3]. Hypoxia¬Ý guarantees proneness of¬Ý Œ≤cells to impairment along with failure in addition to hampering of HIF 1-Œ± actions as well as repression¬Ý of BHLHE40 leads to¬Ý improvement¬Ýof insulin liberation along with hyperglycemia in case of animal models of diabetes, pointing that hypoxia might work¬Ýin the¬Ý form of an innovative therapeutic target for type2diabetes in addition to improvement¬Ý ¬Ýof hypoxia might work¬Ý as advantageous¬Ý for the propagation of¬Ý Œ≤cells impairment in T2D.¬Ý Nevertheless, hypoxia further stimulates ATF3 expression, activation of AMPK inactivation of UPRER¬Ý along with ROS generation (seeFigure5) .
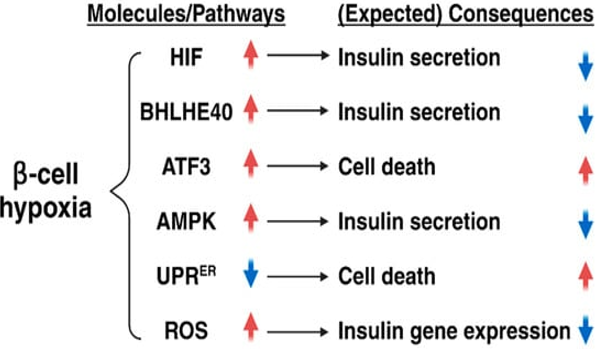
Legend for Figure 5.¬Ý
Courtesy ref no-35-Roles of hypoxia in β-cell function and number. Hypoxia causes impaired insulin secretion through the induction of hypoxia-inducible factor 1 (HIF-1) and basic helix-loop-helix family member E40 (BHLHE40). Hypoxia also suppresses insulin secretion through the activation of adenosine monophosphate-activated protein kinase (AMPK) and the induction of reactive oxygen species (ROS), whereas, it promotes β-cell death via the induction of activating transcription factor 3 (ATF3) and the inhibition of the endoplasmic reticulum unfolded protein response (UPRER).
Moreover, HIF 1-Œ± basically guides the¬Ý reaction to acute hypoxia as well as its expression gets diminished at the time of¬Ýcontinuous hypoxia[65]. Thereby the robustness along with time period¬Ý ¬Ýof¬Ý hypoxia may result¬Ý in activation of¬Ý adaptive reaction in Œ≤cells in a differential manner. Further¬Ý work¬Ý would yield greater insight in the germane aiding of influence of¬Ý every adaptive pathway in the event of manner¬Ý Œ≤cell hypoxia would be¬Ý essential for buttressing our understanding regarding pathophysiological mechanistic¬Ý modes¬Ýof T2 diabetes mellitus . Greater¬Ýwork would aid¬Ý in¬Ý ¬Ýprovision of influence regarding innovative information in¬Ý ¬Ý reference to influence of¬Ý hypoxic stress over Œ≤cells impairment in addition to the efficacy¬Ý of Œ≤cells hypoxia in the¬Ý form of¬Ý ¬Ýantidiabetic therapeutic¬Ý target.¬Ý
Moreover the impairment of Œ≤ cells possess the¬Ý capacity of generating via different mechanistic¬Ý modes, inclusive of OS/ER or hypoxic stress, in addition to through inducing cytokines; such events result¬Ý in apoptosis, unregulated autophagy as well as and do not proliferate. Transdifferentiation amongst Œ≤ cells along with Œ± cells takes place in¬Ýsome pathological situations, in addition to and upcoming¬Ý corroboration pointing that the Œ≤-cell dedifferentiation or transdifferentiation might be responsible for the diminished Œ≤-cell mass found in patients with robust T2DM. FOXO1,portrays¬Ý a crucial¬Ýtranscription factor in insulin signalling(rev in detail by us in ref 20and23,63],. Liang et el. [66],further documented HIF 1-Œ±/ FOXO1axis regulated autophagy conferred¬Ý protection¬Ý for Œ≤cell survival in case of¬Ý hypoxia in human¬Ý islet¬Ýby utility of CoCl2 escalating Œ≤-cell apoptosis as well as choloroquine aggravated autophagy hampering in case of FOXO1KO accelerated apoptosis¬Ý ¬Ýwith immunofluorescent staining¬Ý reported that significant reduction¬Ý in LC3 in addition to¬Ý p62/SQSTMW expression quantities which¬Ý were negatively¬Ý associated¬Ýwith glycated haemoglobin A1c (HbA1c ) inpatients with robust¬Ý T2DM. Thereby HIF 1-Œ±/ FOXO1¬Ý ¬Ýaxis controlled autophagy which is¬Ý of¬Ý advantage¬Ý for Œ≤cells survival under hypoxia in human¬Ý islets. Furthermore,emerging reports¬Ý have displayed advantage¬Ý of restoration of autophagy¬Ý in pancreatic Œ≤cells¬Ý as a therapeutic target for type2Diabetes as¬Ý ¬Ýdisplayed by¬Ý Zhao etal. [67], as well as we had¬Ý detailed autophagy comprehensively previously [68]. Thereby targeting hypoxia as well as autophagy might be the¬Ý next line of treatment for preventing robust T2DM¬Ý ¬Ýthe way illustrated in figure 6¬Ý by different plant extracts and tradional Chinese medicines[rev in det inref no67]¬Ý .¬Ý
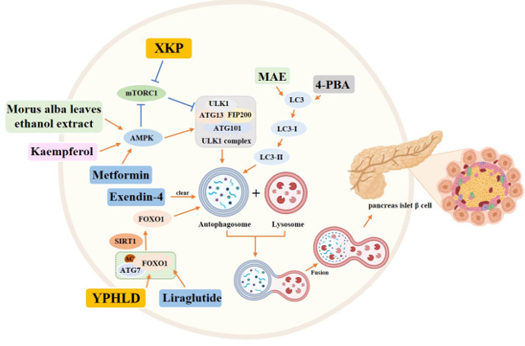
Legend for Figure 6
Courtesy ref no-67-Role of autophagy in pancreatic islet Œ≤ cells in the diabetic state. Yellow rectangle: Traditional Chinese compounds; Blue rectangle: Chemical drugs; Pink rectangle: Monomers from Chinese Herbal; Gray rectangle: Experimental Chemicals.¬Ý‚Üí: activate:¬Ý‚üû: inhibit.
References
-
Ong KL,Stafford LK, McLaughlin SA,Boyko EJ,Vollset SE,Smith AE,etal. Global, regional, and national burden of Diabetes from 1990 to 2021 with projections of Prevalence to2050: a systematic analysis for the global burden of Diabetes study2021. Lancet 2023;402:2354-66.
View at Publisher | View at Google Scholar -
DeFronzo RA, Abdul –Ghani MA. Preservation of βcell functions: the key to Diabetes prevention. J Clin Endocrinol Metab 2011;96:3068-75.
View at Publisher | View at Google Scholar -
Prentki M,NolanCJ. Islets βcells failure in Type2 Diabetes. J Clin Investig 2006;116:1802-12.
View at Publisher | View at Google Scholar -
HudishLI,Reusch JEB,Sussel L. βcelldysfunction during progression of metabolic syndrome to Type2 Diabetes. J Clin Investig 2019; 129:4001-8.
View at Publisher | View at Google Scholar -
Bensellam M,Laybutt DR,Jonas JC. The molecular mechanisms of pancreatic βcells gluotoxicity: recent findings and Future research directions. Mol Cell Endocrinol 2012;364:1-27.
View at Publisher | View at Google Scholar -
Sato Y,EndoH, Okuyama H Takeda, T,Iwahashi H,Imagawa A, YamagataK,et al. Cellular hypoxia pancreatic βcells due to high levels of oxygen consumption for insulin secretion in vitro. J Biol Chem 2011;286:12524-32.
View at Publisher | View at Google Scholar -
Bensellam M,Duvillie B,Rybachuk G,Laybutt DR,Magnan C,Guiot Y,et al. Glucose induced O2 consumption Hypoxia inducible factors-1 and2in rat insulin secreting βcells. .PLoS ONE2012:7:e29807.
View at Publisher | View at Google Scholar -
Catrina SB, ZhengX. Hypoxia and hypoxia inducible factors in Diabetes and its Complications. Diabetologia 2021;64:709-16.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M Sarcopenic Obesity-A Minireview-Does it Lead to a Greater Incident of Type 2 Diabetes, Metabolic Syndrome or Mortality than When Sarcopenia or Obesity Exist Separately. Archives of Diabetes and Endocrine System 2019;(2(1):26-32
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M.. Importance of Simultaneous Treatment of Obesity andDiabetes Mellitus: A Sequelae to the Understanding of Diabesity-A Review. Obes Res Open J. 2019; 6(1): 1-10. doi: 10.17140/OROJ-6-136
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. “Obesity and Oral Health-Emphasis on Early Childhood Caries (ECC) and Trying to ImplementPrevention Strategies Right from Neonatal Age Besides Updating on other Causes of the Same-A Mini Review”. EC Dental Science 2019;18(11); 51-60.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Utilization of Extracellular Vesicles for Treatment of Type 1 Diabetes Mellitus (T1DM) Along with Type 2 Diabetes Mellitus (T2DM) besides Complications Associated with Diabetes- A Systematic Review. J Clin DiabetesObes.2020.1.001-013. DOI: 10.47755/
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Role of Adipocyte impairment in Heart Failure Induction in subjects that are obese along with prediabetes and overt Diabetes mellitus -A Systematic Review.J Cardiol &Card Disord 2021;2(1):1-21.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. ’The Association of Non Viral Liver Diseases from NAFLD to NASH to HCC with the Pandemic of Obesity ,Type 2 Diabetes,or Diabesity & Metabolic Syndrome –Etiopathogenetic Correlation along with Utilization for Diagnostic &Therapeutic Purposes-A Systematic review’’. Journal of Endocrinology Research 2021:3(2):1(1-26). DOI: https://doi.org/10.30564/jer.v3i2.3520
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Potential role of Epigenetic Modulation in prevention or therapy for Diabetic Kidney Disease-still a dream or a reality –A Systematic Review’’.J Diab Nephro Diab Mgmt 2021:1:1(1-26).
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. ‘’Diabetic Cardiomyopathy:An Update on its Pathophysiology with Specific Emphasis on Epigenetics Modifications Besides Treatment-A systematic review’’.BOHR International Journal of Current Research in Diabetes and Preventive Medicine2022; 1 ( 1) : 1–16. https://doi.org/10.54646/bijrdpm.001
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. The utility of phytochemicals obtained from plants for the treatment of type 2 Diabetes Mellitus with Emphasis on the Epigenetic Alterations related to T2DM& their Impact as Therapeutic Agents in the form of so called Epi-drugs:a systematic review. Adv Obes Weight Manag Control. 2021;11(6):195‚Äí206.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M.Role of Trigonella foenum-graecim Extract along with Ursolic Acid a Pentacyclic Triterpenoid as Newer Plant Productsfor the Therapy of Diabetes Mellitus - A Short Communication
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Development of protein tyrosine phosphatase 1B (PTPIB)Inhibitors from marine sources and other natural products-Futureof Antidiabetic Therapy : A Systematic ReviewKorean Journal of Food & Health Convergence 5(3),pp.21-33. ISSN: 2586-7342 © 2019 KFHCA. http://www.kjfhc.or.krdoi: http://dx.doi.org/10.13106/kjfhc.2019.vol5.no3.21
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. ’An Update on the Therapeutic Potential of Herbal Preparations with regards to Molecular & Biochemical Mechanisms in the Management of Diabetes Mellitus :a Systematic Review ‘’. World Journal of Advance Healthcare Research 2022; 6( 3) : 1–17.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Utilization of extracellular vesicles for treatment of Type 1 Diabetes Mellitus ( T1DM) along with 2 T2DM besides Complications associated with Diabetes-A Systematic Review’’. J Clin DiabetesObes;2020(1.)001-013. DOI: 10.47755
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Association of Iron Metabolism Abnormalities as Etiopathogenetic Factor in Alteration of Beta Cell Function and Impairment in Generation of Diabetes Mellitus: A Systematic Review. J Clinical Research and Reports, 2022;11(1); DOI:10.31579/2690-1 919/241.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. An Update onMechanistic Modes in AGEs Stimulated & ERand Inflammatory Stress- Modulated Control of the GLUT4 expression (SLC2A4 promoted) andAtherogenesis in Diabetes Mellitus-A Narrative Review. Mathews J Cytol Histol. 6(1):21:1-25.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. An update of use of therapeutic targeting of macrophages polarization status in the treatment of obesity induced insulin resistance ,chronic inflammation and type2 Diabetes mellitus-A Narrative Review’’. World Journal of Advance Healthcare Research 2023;7(1):1-19.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. ’ Update on Etiopathogenesis of Type 1 Diabetes (T1D):Emphasis on part of Crosstalk of Gut Microbiome, Pancreatic Cells& Bystander Activation of Memory CD8+T cells with Mitochondrial MelatonergicPathway: Treatment Repercussions-A Narrative Review’’. Universal Library of Medical and Health Sciences 2023;1(1):41-64.
View at Publisher | View at Google Scholar -
Rorsman P,Renstrom E. Insulin granule dynamics in pancreatic βcells. Diabetologia 2003;46:1029-45.
View at Publisher | View at Google Scholar -
WangU,Upshaw L,Strong DM,RobertsonRP,Reems JA.Increased oxygen consumption rates in response to high glucose detected by a novel oxygen biosensor system in non human primate and human islets. J Endocrinol 2005;185;445-55.
View at Publisher | View at Google Scholar -
Carlsson PO,Anderson A,Jansson L . Pancreatic isletblood flow in normal and obese hyperglycemic (ob/ob) mice. Am J Physiol Endocrinol Metab 1996;271:E990-E995.
View at Publisher | View at Google Scholar -
Spencer JA, Ferraro F,Roussakis E,KleinA,WuJ,Runnels JM. Direct measurements of local oxygen Concentration in the Bone marrow of live animals. Nature 2014;508:269-73.
View at Publisher | View at Google Scholar -
Carlsson PO,LissP, Anderson A,Jansson L. Measurements of oxygen tension in native and transplanted rat pancreatic islets . Diabetes1998;47:1027-32.
View at Publisher | View at Google Scholar -
Solaini G,Baracca A,Lenaz G,Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys ActaBioenerg 2010; 1797:1171-77.
View at Publisher | View at Google Scholar -
Sato Y, Inoue M,YoshizawaT, YamagataK.Moderate hypoxia induces pancreatic βcell dysfunction with HIF 1 independent gene expression .PLoS ONE2014:9:e1148683.
View at Publisher | View at Google Scholar -
TsuyamaT, Sato Y, YoshizawaT,MatsuokaT YamagataK. Hypoxia causes βcell dysfunction and impairs insulin secretion by activating the transcriptional repressor BHLHE40.EMBORep 2023;24:e56227.
View at Publisher | View at Google Scholar -
YamagataK, TsuyamaT, Sato Y. Roles of βcell function in the progression of Type2 Diabetes. Int J Mol Sci 2024;25:4186.
View at Publisher | View at Google Scholar -
Bensellam M, Jonas JC, Laybutt DR. Mechanisms of βcells dedifferentiation in Diabetes : recent findings and Future research directions. J Endocrinol 2018;236; R109- R143.
View at Publisher | View at Google Scholar -
LeeP,Chandel NS,Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 2020;21:268-83.
View at Publisher | View at Google Scholar -
Keith B, JohnsonRS,Simon MC. HIF1α and HIF 2- α:sibling rivalry in tumor growth and progression. Nat Rev Cancer 2011; 12: 9-22.
View at Publisher | View at Google Scholar -
HoangM,J E,Janssen SM,Nasteska D,Cuozzo F,Hodson DJ,et al. Isoform specific roles of prolyl hydroxylases in the regulation of pancreatic βcell function. Endocrinology 2022;163:bqab226.
View at Publisher | View at Google Scholar -
ChengK,HoK,StokesR,ScottC,Lau SM,Hawthorne WJ,et al. Hypoxia inducible factor-1α regulates βcell function in mouse and human islets. 2010; 120:2171-83.
View at Publisher | View at Google Scholar -
Guillam MT,Hummler E,Schaerer E,WuJY,Birnbaum MJ,Beermann F,et al. Early Diabetes and abnormal postnatal pancreatic development in mice lackingGlut2. Nat Genet 1997;17:327-30.
View at Publisher | View at Google Scholar -
Matchinsky FM .Glucokinase, glucose homeostasis , and Diabetes mellitus. Curr Diabetes Rep 2005;5:171-6.
View at Publisher | View at Google Scholar -
Nomoto H,Pei L,Montemurro C,Rosenburger M,Furterer A,Coppola G,et al. Activation of the HIF1 α /PFKB3 stress response pathway in beta cells in type1 diabetes . Diabetologia 2021;64:709-16.I
View at Publisher | View at Google Scholar -
GuntonJE,Kulkarni RN,YimSH, Okada T, Hawthorne WJ,TsengYH,RobertsonRP,et al.Loss of ARNT /HIF1β mediated altered gene expressionin human Type2 Diabetes. Cell 2005;122:337-49.
View at Publisher | View at Google Scholar -
ZhengX,Naraynan S, XuC,Angelstig SE,Grunler J, ZhaoA,et al. Repressionof Hypoxia inducible factor-1α contributes to increased reactive oxygen species production in Diabetes. eLife 2022;11:e70714.
View at Publisher | View at Google Scholar -
Ilegems E,Bryzgalova G,Correia J,Yesildag B,Berra E,Ruas JL,et al. HIF1α inhibitor PX-478 preserves pancreatic βcells function in Diabetes. Sci Transl Med 2022; 14:eaba9112.
View at Publisher | View at Google Scholar -
CantleyJ,Selman C,Shukla D,Abramov AY,Forstreuer F,Esteban MA,et al. Deletion of the vonHippel-Lindau gene in pancreatic βcells impairs glucose homeostasis in mice. J Clin Investig 2009; 119:125-35.
View at Publisher | View at Google Scholar -
SemenzaGL. HIF1 mediates responses to intratumoral hypoxia and oncogenic mutations .J Clin Investig 2013; 123:3664-71.
View at Publisher | View at Google Scholar -
MaechlerP,WolheimCB. Mitochondrial function in normal and diabetic βcells. Nature 2001;414: 807-12.
View at Publisher | View at Google Scholar -
MoonJS,Riopel M,Seo JB,Herrero AguayoV,Isaac R,LeeYS. HIF2 α preserves mitochondrial activity and glucose sensing in compensating βcells in Obesity. Diabetes2022;71:1508-24.
View at Publisher | View at Google Scholar -
ScortegagnaM, DingK,O Y,G A,T F,YanLJ,et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas 1-/- mice. Nat Genet 2003;35:331-40.
View at Publisher | View at Google Scholar -
Cavadas MAS, CheongA,TaylorCT.The regulation of transcriptional repression in hypoxia. Exp Cell Res 2017;356:173-81.
View at Publisher | View at Google Scholar -
Gerber PA,Bellomo EA,Hodson DJ,Meur G,Solomou A,Mitchell RK,et al. Hypoxia lowers SLC30A8/ZnT8 expression and free cytosolic Zn2+ in pancreatic βcells . Diabetologia 2014;57:1635-44.
View at Publisher | View at Google Scholar -
SatoF,Bhawal UK,Yoshimura T,Muragaki Y.DEC 1 and DEC2 crosstalk between circadian rhythm and tumor progression. J Cancer 2016; 7: 153-9.
View at Publisher | View at Google Scholar -
Cataldo LR,S T,Achanta K,Bharat S,Prasad RB,et al. MAFA and MAFB regulate exocytosis related genes in human βcells. Acta Physiol 2022;234:e13761.
View at Publisher | View at Google Scholar -
Puigserver P.Spiegelman BM. Peroxisome Proliferator Activated Receptor γCoactivator -1α(PGC-1α): transcriptional coactivator and metabolic regulator . Endocrin Rev 2003;24:78-90.
View at Publisher | View at Google Scholar -
Zmuda EJ,QiL, ZhuMX,Mirmira RG,Montminy MR,Hai T.The role of ATF3an adaptive repressor gene in high fat diet induced Diabetes and pancreatic βcells dysfunction. Mol Endocrinol 2010;24:1423-33.
View at Publisher | View at Google Scholar -
KuHC, Cheng CF.Master regulator activating transcription factor 3in metabolic homeostasis and cancer. Front Endocrinol 2020;11:556.
View at Publisher | View at Google Scholar -
Garcia D,Shaw RJ,AMPK: mechanisms ofcellular energy sensing and restoration of metabolic balance. Mol Cell 2017;66:789-800.
View at Publisher | View at Google Scholar -
MiuraA,YamagataK,KakaeiM,HatakeyamaH,TakayashiN,Fukui K,et al. Hepatocyte nuclear factor 4 alpha is essential for glucose stimulated insulin secretionby pancreatic βcells. J Biol Chem 2006;281:5246-57.
View at Publisher | View at Google Scholar -
Sato Y, TsuyamaT, Sato C,KarimMF, YoshizawaT, Inoue M, YamagataK. Hypoxia reduces HNF-4α/MODY1 protein expression in pancreatic βcells by activating AMP)-activated protein kinase . J Biol Chem 2017;292: 8716-28.
View at Publisher | View at Google Scholar -
Metcalf MG,Higushi-Sanabria R,GarciaG,Kimberly Tsui C,Delin A.Beyond the cell factory: homeostatic regulation of and by the UPRER . Sci Adv 2020;6:eabg9614 .
View at Publisher | View at Google Scholar -
Bensellam M,Maxwell EL,ChanJY,Luzuriags J,West PK,Jonas JC,GuntonJE,Laybutt DR. Hypoxia reduces ER to golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mousebeta cells . Diabetologia 2016;59:1459-502.
View at Publisher | View at Google Scholar -
Kitamura T. The role of FOXO1 βcell failure and type2Diabetes mellitus. Nat Rev Endocrinol 2013; 9:615-23.
View at Publisher | View at Google Scholar -
SemenzaGL. Hypoxia inducible factors:coupling glucose metabolism andredox regulation with induction of the breast cancer stem cell phenotype. EMBO J 2017; 36: -252-9.
View at Publisher | View at Google Scholar -
HuY,LuH,LiH,GeJ.M basis and clinical implications of HIF1s in cardiovascular diseases. Transl Mol Med 2022;28:916-38.
View at Publisher | View at Google Scholar -
LiangR, Liu N, CaoJ,LiuT,SunP,CaiX,et al. HIF 1-α/ FOXO1axis regulated autophagy is protective for βcells survival under hypoxia in human islets . Biochim Biophys Acta Mol Basis Dis 2022;1868:166356.
View at Publisher | View at Google Scholar -
Zhao X,BieLY,PangDR,LiX,YangLF, ChenDD,et al. The role of autophagy in the treatment of typeII Diabetes mellitus and its complications:a review. Front Endocrinol(Lausanne) 2023;14:1228045.
View at Publisher | View at Google Scholar -
Kulvinder Kochar Kaur,Allahbadia GN,Singh M. Mode of Actions of Bile Acids in Avoidance of Colorectal Cancer Development and Therapeutic Applications in Treatment of Cancers--A Narrative Review’’. Journal of Pharmacy and Nutrition Sciences, 2022, 12, 35-53.
View at Publisher | View at Google Scholar







